
НОВАЯ ЭНДОНУКЛЕАЗА РЕСТРИКЦИИ LmnI УЗНАЕТ И РАСЩЕПЛЯЕТ НЕПАЛИНДРОМНУЮ ПОСЛЕДОВАТЕЛЬНОСТЬ ДНК 5’-GCTCC(1/-1)-3’
Чернухин В.А., Гончар Д.А., Абдурашитов М.А., Наякшина Т.Н., Дедков В.С., Михненкова Н.А., Ломаковская Е.Н., Дементьев С.Н., Дегтярев С.Х.
В.А. Чернухин, Д.А. Гончар, М.А. Абдурашитов, Т.Н. Наякшина, В.С. Дедков, Н.А. Михненкова, Е.Н. Ломаковская, С.Н.Дементьев1, С.Х. Дегтярев
Выделенный нами из природных изолятов бактериальный штамм Lysinibacillus manganicus An22 является продуцентом эндонуклеазы рестрикции LmnI, узнающей непалиндромную последовательность ДНК 5’-GCTCC-3’/3’-CGAGG-5’. Данная последовательность представляет собой новый прототип сайтов узнавания рестриктаз. Препарат эндонуклеазы рестрикции LmnI с концентрацией 1000 ед/мл был получен путем очистки в четыре хроматографические стадии. Показано, что новый фермент гидролизует ДНК с образованием 3’-выступающих “липких” двухнуклеотидных концов как показано стрелками — 5’-GCTCCN↓-3’/3’-CGAG↑GN-5’, и относится к подтипу IIS эндонуклеаз рестрикции.
ООО «СибЭнзайм», 630117, Новосибирск, Россия
1 — Институт Геологии и Минералогии им. В.С. Соболева СО РАН
ВВЕДЕНИЕ
Сайт-специфические эндонуклеазы рестрикции (рестриктазы, ЭР) вместе с соответствующими ДНК-метилтрансферазами представляют собой бактериальную систему рестрикции-модификации. Наиболее изученными и востребованными в молекулярно-биологических исследованиях являются рестриктазы типа II, они нашли широкое применение в картировании протяженных ДНК, клонировании генов, ДНК-диагностике и т.д. Эндонуклеазы рестрикции второго типа узнают короткие последовательности в двуцепочечной ДНК, размерами от четырех до восьми пар нуклеотидов, и специфически расщепляют их либо внутри сайта узнавания, либо на фиксированном расстоянии вблизи от этого сайта. При этом, вторые ферменты узнают асимметричную (непалиндромную) последовательность ДНК и согласно современной классификации [1] относятся к подтипу IIS (от англ. shift). На текущий момент известно более 90 подобных ферментов, узнающих уникальные асимметричные сайты и являющихся прототипами [2].
Данная работа посвящена установлению субстратной специфичности эндонуклеазы рестрикции LmnI, выделенной из бактериального штамма Lysinibacillus manganicus An22 и являющейся новым прототипом.
МАТЕРИАЛЫ И МЕТОДЫ
В работе использовались реактивы производства “Sigma” (США), “Fisher” (США), “Panreac” (Испания) и “Хеликон” (Россия). Для хроматографической очистки фермента применялись следующие носители: фенил-сефароза (“Sigma”, США), гидроксиапатит и биогель A-0,5m (“BioRad”, США). Наработка биомассы штамма-продуцента осуществлялась c использованием компонентов питательной среды фирмы ”Organotechnie” (Франция).
Препараты ферментов, ДНК, дезоксинуклеозидтрифосфаты и синтетические олигонуклеотиды, а также маркёры молекулярного веса ДНК 1 kb, использованные в работе, произведены в ООО «СибЭнзайм» (Россия). При определения места гидролиза межнуклеотидных связей использовали γ[32P]-меченый ДНК-дуплекс. Исходные олигодезоксирибонуклеотиды метили с помощью Т4-полинуклеотидкиназы и γ[32P]-АТФ. Для разделения продуктов ферментативного гидролиза меченых олигонуклеотидов проводили электрофорез в 20% ПААГ, содержавшем 7 М мочевину.
Морфологические и физико-биохимические свойства штамма изучали с использованием известных методик [3]. Определение вида микроорганизма проводили по определителю Берджи [4] и по результатам секвенирования фрагмента гена 16S рРНК.
Выделение плазмидной ДНК pBR322 [5] осуществляли с использованием набора “QIAGEN GmbH” (Германия) согласно протоколам производителя.
Определение нуклеотидной последовательности ДНК ПЦР-фрагмента гена 16S рРНК осуществляли методом Сэнгера на автоматическом секвенаторе ABI 3130xI Genetic Analyzer (“Applied Biosystems”, США) согласно протоколам производителя.
Расчет теоретических картин гидролиза ДНК-субстратов с известной последовательностью производили с помощью программы Vector NTI Suite 7.
Выращивание клеток штамма L. manganicus An22.
Клетки штамма Lysinibacillus manganicus An22 выращивали в термостатированнной качалке при 30 оС в 0,5-литровых колбах, содержащих по 0,3 л питательной среды LB (1% триптон, 0.5% дрожжевой экстракт, 0.5% NaCl, pH 7.6) с добавлением 0.1% MgSO4 и 0.001% тиамина при перемешивании со скоростью 120 об/мин. Культура росла в течение 18-20 часов до оптической плотности A550=1,9±0,3. Клетки собирались центрифугированием при 8000 g в течение 15 мин на центрифуге J2-21 “Beckman” (США). Биомассу хранили при -20 оС.
Тестирование рестриктазной активности Lmn I.
Для тестирования активности рестриктазы LmnI 1 мл выросшей культуры переносили в 1.5-мл пробирки “Eppendorf” и центрифугировали при 6000 g в течение 2 мин на микроцентрифуге “Eppendorf 5804” (Германия). Осадок суспендировали в 180 мкл воды, и готовили грубый лизат клеток по методу, описанному ранее [6]. Затем аликвоту грубого лизата 5 мкл добавляли к 20 мкл реакционной смеси, содержавшей 1 мкг ДНК фага лямбда в SE-буфере «B» (10 мМ Трис-HCl (pH 7.6), 10 мМ MgCl2, 1 мМ ДТТ), и ставили серию разведений в 4 и 16 раз. Смесь инкубировали в термостате на 37 оС в течение 1 ч. Продукты реакции наносили на 1% агарозный гель и проводили электрофорез в Трис-ацетатном буфере (50 мМ Трис-ацетат (pH 8.0), 20 мМ ацетата натрия, 2 мМ ЭДТА) при напряжении 150 V. После окрашивания бромистым этидием гель фотографировали в УФ-свете.
При тестировании активности ЭР LmnI в хроматографическом профиле аликвоты по 1 мкл из фракций добавляли к 20 мкл реакционной смеси, полученную смесь инкубировали 10 мин при 37 оС, наносили на 1% агарозный гель и проводили электрофорез в Трис-ацетатном буфере.
Для определения последовательности ДНК, узнаваемой ЭР LmnI, 2 мкл препарата фермента добавляли к 20 мкл реакционной смеси, содержащей 1 мкг субстратной ДНК и однократный реакционный SE-буфер «B». Смесь инкубировали при 37 оС в течение 2 ч, после чего аликвоты по 10 мкл наносили на 1% агарозный гель и проводили электрофорез в трис-ацетатном буфере. Полученные картины гидролиза сравнивали с теоретически рассчитанными.
Определение позиции гидролиза ДНК для фермента LmnI.
В качестве субстрата для определения позиции гидролиза ферментом Lmn1 на верхней и нижней цепи ДНК мы использовали олигонуклеотидный дуплекс, состоящий из взаимно комплементарных дезоксирибоолигонуклеотидов Lmn1d и Lmn2r длиной 38 нуклеотидов каждый:
Lmn1d: 5’-CCCTTTCCTCTGCCCGCGGAGCTTGATGATGCTTTCCC-3’
Lmn2r: 5’-GGGAAAGCATCATCAAGCTCCGCGGGCAGAGGAAAGGG-3’
Данный дуплекс содержит предположительный сайт узнавания LmnI и сайты узнавания нескольких контрольных ферментов, в том числе MnlI, SfaNI и Sfr303I. При постановке реакции с ЭР LmnI и контрольными ферментами дуплекс предварительно модифицировали путем включения в верхнюю или нижнюю цепь радиоактивной метки. При этом нужную цепь метили с 5’-конца с помощью Т4-полинуклеотидкиназы и γ[32P]-АТФ, после чего очищали от побочных продуктов реакции путем гель-фильтрации на Sephadex G-50, добавляли комплементарную немеченую цепь, пробирку с реакционной смесью прогревали 5 мин при 95 оС, а затем охлаждали на рабочем столе до комнатной температуры. Реакционная смесь объемом 20 мкл содержала SE-буфер «B», меченый олигонуклеотидный дуплекс в концентрации 65 нМ и 2 мкл эндонуклеазы рестрикции. Смесь инкубировали в течение 1 ч при температуре 37 оС, после чего аликвоты по 10 мкл наносили на 20% ПААГ с 7 М мочевиной и проводили электрофорез в трис-боратном буфере. Радиоавтографию геля проводили с помощью прибора Cyclone Storage System (Packard Instrument Co., США).
Получение препарата фермента LmnI.
Выделение фермента проводили при 4 оС с использованием растворов:
буфер А — 20 мМ Трис-HCl pH 7.6 и 7 мМ меркаптоэтанол;
буфер Б — 10 мМ К-фосфат pH 7.2 и 7 мМ меркаптоэтанол;
буфер В — 50 мМ Трис-HCl pH 7.6 и 7 мМ меркаптоэтанол.
Экстрагирование. 20 г биомассы суспендировали в 60 мл буфера А с добавлением 0,2 М NaCl, 1 мМ фенилметилсульфонилфторида, 0,1 мг/мл лизоцима, 0,1% Тритона Х-100. Клетки разрушали ультразвуком на дезинтеграторе Soniprep 150 (“MSE”, Англия) с диаметром адаптера 2 см. Обработку проводили при амплитуде 20 мкм 10 раз по 1 мин с интервалами по 1 мин, охлаждая суспензию в ледяной бане. Экстракт осветляли при 15000 об/мин в течение 30 мин в роторе JA-20 на центрифуге J2-21 (“Beckman”, США).
Суммарный белок экстракта осаждали добавлением сульфатом аммония до 60% и центрифугировали при 12000 об/мин в течение 20 мин. Осадок растворяли в 60 мл буфера В.
Гель-фильтрация. Полученную фракцию объемом 60 мл наслаивали на колонку с биогелем А-0,5m (“Bio-Rad”, США, V=500 мл), уравновешенную буфером А с 0,4 М NaCl, 0,1% Тритоном Х-100, и промывали этим же буфером со скоростью 30 мл/ч. Собирали 50 фракций по 10 мл. По результатам анализа фракции, содержащие целевую активность объединяли и осаждали добавлением сульфатом аммония до 60%. Смесь центрифугировали 40 мин при 12000 об/мин. Затем осадок растворяли в 25 мл буфера В.
Хроматография на гидроксиапатите. Полученную фракцию диализовали против 2 л буфера А в течение 16 часов, наносили на колонку с гидроксиапатитом объемом 20 мл, уравновешенную буфером Б и промывали двумя колоночными объемами того же буфера. Сорбированный материал вымывали линейным градиентом К-фосфатного буфера, рН 7.2, от 0,04 до 0,25 М К-фосфата объемом 350 мл. Из 60 фракций объединили пробирки под номерами 39-59 (0,18 М К-фосфат), содержащие целевую активность.
Хроматография на фенил-сефарозе. К полученной фракции добавляли 1 М Трис-НСl, pH 7.6, до концентрации 60 мМ, глицерин до концентрации 5%, 4 М NaCl до концентрации 0,8 М, сухой сульфат аммония до концентрации 1,7 М и наносили на колонку с фенил-сефарозой объемом 4 мл, уравновешенную буфером В, содержащим 1,7 М сульфат аммония. Промывали 2 колоночными объемами того же буфера. Сорбированный материал вымывали линейным градиентом сульфата аммония от 1 до 0 М в буфере В, объемом 160 мл. Из 40 фракций объединяли пробирки под номерами № 29-39 (0,12 M сульфат аммония), содержащие целевой фермент.
Рехроматография на гидроксиапатите. Полученную фракцию диализовали против 1 л буфера А в течение 16 часов и наносили на колонку с гидроксиапатитом объемом 1,5 мл, уравновешенную буфером Б, промывали двумя колоночными объемами буфера Б. Сорбированный материал вымывали линейным градиентом К-фосфатного буфера, pH 7.2, от 0,04 до 0,25 М К-фосфата объемом 100 мл. Из 40 фракций объединяли пробирки под номерами № 15-18, содержащие пик активности LmnI.
Концентрирование и хранение препарата фермента. Полученную фракцию диализовали в течение 20 ч против 300 мл буфера А, содержащего 0,25 М NaCl и 50% глицерин, и хранили при -20 оС. За одну единицу активности принимали количество фермента, необходимое для исчерпывающего расщепления 1 мкг ДНК фага лямбда в 50 мкл реакционной смеси за 1 ч при 37оС.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В ходе массового скрининга бактериальных штаммов, выделенных из проб почвы и пресной воды, на предмет наличия эндонуклеаз рестрикции был обнаружен штамм, первоначально обозначенный как An22, продуцирующий фермент с неизвестной специфичностью. Образцы почвы и пресной воды были отобраны и предоставлены ведущим инженером ИГМ СО РАН С.Н. Дементьевым. В результате проведенных микробиологических исследований и секвенирования фрагмента гена 16S рРНК обнаруженный штамм бактерий был определен как Lysinibacillus manganicus An22, а эндонуклеаза рестрикции из него была названа LmnI согласно общепринятой номенклатуре [7].
В результате наработки культуры клеток штамма L. manganicus An22 было получено 20 г биомассы (4 г/л) с удельной активностью 8-10 ед/г. После четырехстадийной хроматографической очистки из данного количества биомассы было получено 2,5 мл препарата эндонуклеазы рестрикции LmnI с концентрацией 1000 ед/мл.
Оптимальными условиями для работы фермента оказались SE-буфер «B» и температура 37оС (данные не приведены). Полная инактивация ЭР LmnI в реакционной смеси происходила при нагревании до 65оС за 20 мин.
Определение специфичности рестриктазы LmnI проводили, используя в качестве субстратов ДНК фагов λ и Т7, а также ДНК плазмиды pBR322. Ниже приведены картины гидролиза этих ДНК ферментом LmnI, полученные экспериментально (рис. 1а), и рассчитанные теоретически картины расщепления этих же ДНК по сайту 5’-GCTCC-3’/3’-CGAGG-5’ (рис. 1б). Видно, что длины экспериментально полученных фрагментов ДНК совпадают с расчетными.
Рисунок 1.
Определение специфичности эндонуклеазы рестрикции LmnI на ДНК фагов λ, Т7 и плазмиды pBR322.
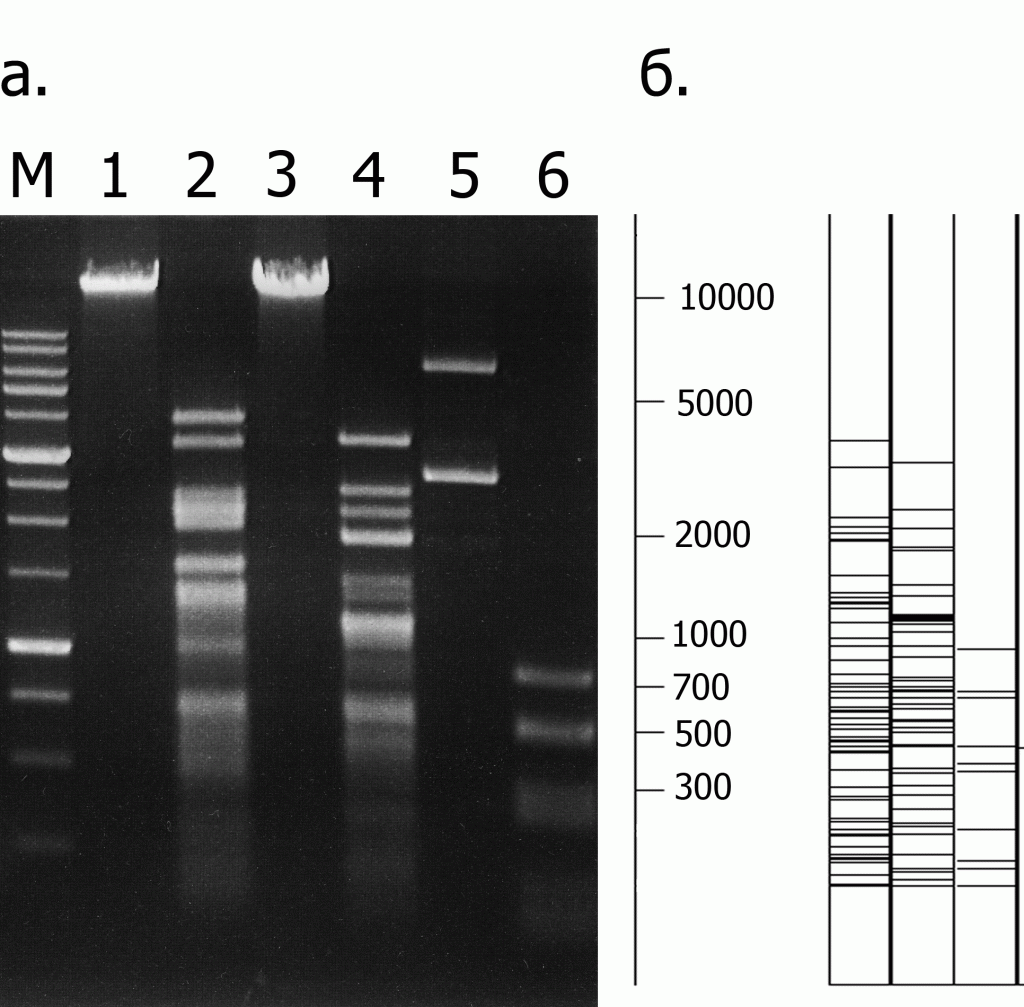
а. Экспериментально полученная картина гидролиза ферментом LmnI трех субстратных ДНК. Дорожки: 1 – ДНК фага λ, 2 – ДНК λ + LmnI, 3 – ДНК фага Т7, 4 – ДНК Т7 + LmnI, 5 – ДНК плазмиды pBR322, 6 – pBR322 + LmnI, М – 1 kb ДНК-маркеры молекулярного веса (от 0,25 до 10 т.п.н.). Продукты расщепления разделены в 0,8 % агарозном геле.
б. Теоретически рассчитанная картина гидролиза ДНК фагов λ, Т7 и плазмиды pBR322 по сайту 5’-GCTCC-3’/3’-CGAGG-5’. Слева приведена шкала длин фрагментов ДНК, выраженная в парах оснований.
Чтобы подтвердить последовательность узнавания для ЭР LmnI и определить место гидролиза ДНК новым ферментом мы использовали олигонуклеотидный дуплекс, состоящий из дезоксирибоолигонуклеотидов Lmn1d и Lmn2r. Последовательность нуклеотидов в дуплексе была подобрана таким образом, чтобы она включала сайт узнавания исследуемого фермента LmnI, а также сайты узнавания нескольких контрольных ЭР с известной позицией гидролиза. Поскольку LmnI узнает асимметричный сайт, то исходя из литературных данных, с высокой вероятностью можно было предположить, что позиции гидролиза ферментом верхней и нижней цепи ДНК тоже окажутся несимметричными относительно центра узнаваемой последовательности нуклеотидов.
На рисунке 2 показана структура дуплекса Lmn1d/Lmn2r, отмечены сайты узнавания LmnI и ферментов, взятых в качестве контрольных – SfaNI (5’-GCATC(5/9)-3’), Sfr303I (5’-CCGC↓GG-3’) и MnlI (5’-CCTC(7/6)-3’). Стоит отметить, что сайты узнавания ЭР Sfr303I и LmnI перекрываются на два нуклеотидных остатка (рис. 2). При этом Sfr303I расщепляет верхнюю цепь дуплекса с 5’-конца перед сайтом LmnI, а нижнюю цепь – за два нуклеотида до 3’-конца сайта LmnI. В свою очередь, узнаваемые последовательности SfaNI и MnlI находятся на удалении от сайта LmnI, но их позиции гидролиза ДНК оказываются либо внутри последовательности, узнаваемой LmnI (SfaNI, верхняя цепь), либо непосредственно вблизи этой последовательности (MnlI, верхняя и нижняя цепи).
Рисунок 2.
Структура дуплекса Lmn1d/Lmn2r
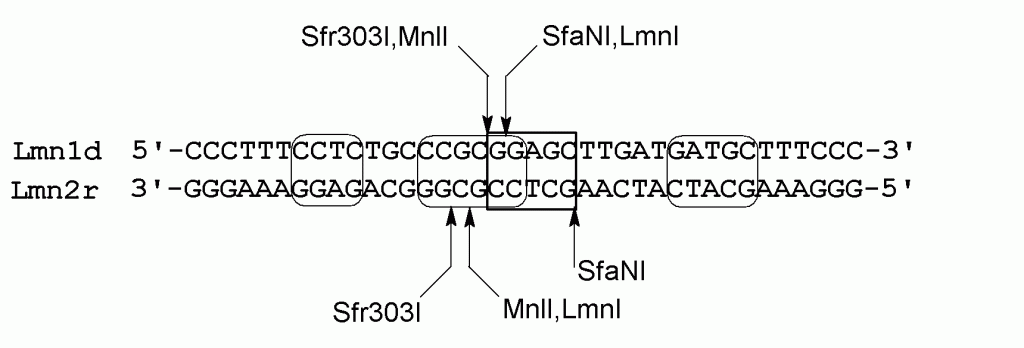
Сайт узнавания LmnI выделен прямоугольной рамкой. Сайты узнавания контрольных ферментов MnlI, SfaNI, Sfr303I выделены закруглёнными рамками. Места расщепления верхней и нижней цепи обозначены стрелками.
В серии проведенных экспериментов верхнюю либо нижнюю цепь дуплекса метили с 5’-конца как описано в разделе «Материалы и методы». Далее осуществляли гидролиз этого дуплекса ЭР LmnI, контрольными ферментами, и после проведения электрофореза в денатурирующем ПААГ, сравнивали полосы, соответствующие продуктам гидролиза.
На рисунке 3 представлены радиоавтографы продуктов гидролиза дуплекса Lmn1d*/Lmn2r и Lmn1d/Lmn2r* рестриктазой LmnI и соответствующими контрольными ферментами после проведения электрофореза в денатурирующем ПААГ.
Рисунок 3.
Определение позиции расщепления ДНК эндонуклеазой рестрикции LmnI с использованием γ[32P]-меченого олигонуклеотидного дуплекса.
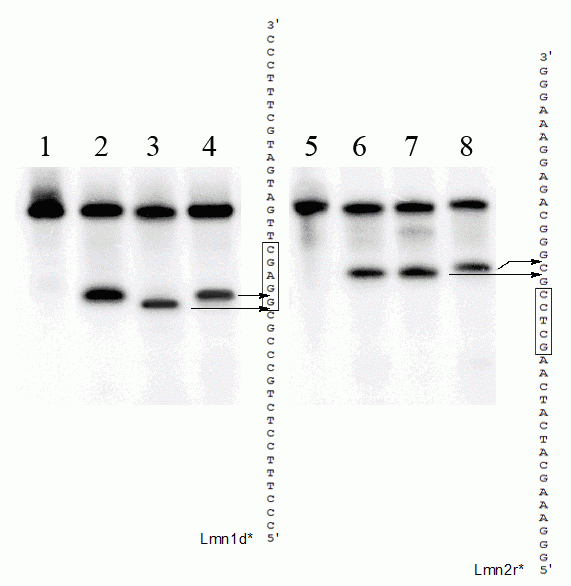
Дорожки: 1 – исходный дуплекс Lmn1d*/Lmn2r; 2 — Lmn1d*/Lmn2r + LmnI; 3 — Lmn1d*/Lmn2r + Sfr303I; 4 — Lmn1d*/Lmn2r + SfaNI; 5 — исходный дуплекс Lmn1d/Lmn2r*; 6 — Lmn1d/Lmn2r* + MnlI; 7 — Lmn1d/Lmn2r* + LmnI; 8 — Lmn1d/Lmn2r* + Sfr303I. Меченая цепь обозначена *. Последовательность узнавания LmnI выделена рамкой. Продукты разделены в 20 % ПААГ с 7 М мочевиной.
Как видно из рисунка 3 продукты гидролиза дуплекса Lmn1d*/Lmn2r, содержащего меченую верхнюю цепь, совпадают для ЭР LmnI и SfaNI (дорожки 2 и 4, соответственно), а полоса, соответствующая продукту расщепления Sfr303I смещена на одну позицию вниз относительно полос LmnI и SfaNI (дорожка 3). Следовательно, последовательность 5’-GGAGC-3’ расщепляется ферментом LmnI в районе между первым и вторым остатками гуанина: 5’-G↓GAGC-3’.
Из рисунка 3 можно также увидеть, что продукты гидролиза дуплекса Lmn1d/Lmn2r*, содержащего меченую нижнюю цепь, совпадают для ЭР MnlI и LmnI (дорожки 6 и 7, соответственно), а полоса, соответствующая продукту расщепления Sfr303I смещена на одну позицию вверх относительно полос MnlI и LmnI (дорожка 8). Следовательно, фермент LmnI расщепляет последовательность 5’-GCTCC-3’ на расстоянии одного нуклеотида от 3’-конца – 5’-GCTCCg↓-3’.
Из представленных данных можно сделать вывод, что новый фермент узнает непалиндромную последовательность ДНК 5’-GCTCCN↓-3’/3’-CGAG↑GN-5’ и гидролизует её, как показано стрелками с образованием 3’-выступающих “липких” двухнуклеотидных концов. Поскольку на текущий момент не обнаружено других рестриктаз, умеющих такую же последовательность узнавания, фермент LmnI можно считать новым прототипом. Ранее мы предположили, что активный центр фермента, участвующий в расщеплении ДНК, является более консервативным, чем узнаваемая последовательность, что позволяет выделить целые группы рестриктаз, имеющие общий механизм расщепления ДНК [8]. В работе [8] мы предложили схему расположения узнаваемых последовательностей и мест расщепления ДНК для группы из 5-ти ферментов, образующих 3’-выступающие двухнуклеотидных концы. В базе данных 2019 года [2] указаны уже 7 ферментов (включая LmnI), имеющих асимметричный сайт узнавания и расщепляющих ДНК с образованием 3’-выступающих двухнуклеотидных концов. Помимо LmnI это ферменты BsmI (5’-GAATGC(1/-1)-3’), BsrI (5’-ACTGG(1/-1)-3’), BsrDI (5’-GCAATG(2/0)-3’), BtsI (5’-GCAGTG(2/0)-3’), BstF5I (5’-GGATG(2/0)-3’) и BtsMutI (5’-CAGTG(2/0)-3’). Схема расположения сайтов узнавания и точек расщепления ДНК для этих семи ферментов представлены в таблице 1.
Таблица 1
Схема расположения сайтов узнавания и точек расщепления ДНК для семи ферментов, имеющих асимметричный сайт узнавания и расщепляющих ДНК с образованием 3’-выступающих двухнуклеотидных концов. Полностью совпадающие нуклеотидные остатки подчеркнуты| Эндоноклеаза рестрикции | Сайт узнавания верхней цепи | Сайт узнавания нижней цепи |
|---|---|---|
| LmnI | 5’-GCTCCN↓-3’ | 5’-NG↑GAGC-3’ |
| BsmI | 5’-GAATGCN↓-3’ | 5’-NG↑CATTC-3’ |
| BsrI | 5’-ACTGGN↓-3’ | 5’-NC↑CAGT-3’ |
| BsrDI | 5’-GCAATGNN↓-3’ | 5 ’-NN↑CATTGC-3’ |
| BtsI | 5’-GCAGTGNN↓-3’ | 5’-NN↑CACTGC-3’ |
| BtsMutI | 5’-CAGTGNN↓-3’ | 5’-NN↑CACTG-3’ |
| BstF5I | 5’-GGATGNN↓-3’ | 5’-NN↑CATCC-3’ |
Как видно из таблицы все представленные ферменты имеют общий элемент в структуре сайта узнавания 5’-RNTSNN↓-3’/-3’YNAS↓NN-5’, где R означает A или G, Y – T или C, а S — G или C. Таким образом, эндонуклеазы рестрикции типа IIS, расщепляющие ДНК с образованием 3’-выступающих двухнуклеотидных концов, имеют сайт узнавания размерами 5-6 нуклеотидов и содержат последовательность нуклеотидов 5’- RNTS — 3’.
Новая эндонуклеаза рестрикции LmnI из бактериального штамма Lysinibacillus manganicus An22, узнает последовательность ДНК 5’-GCTCC(1/-1)-3’. LmnI относится к подтипу IIS эндонуклеаз рестрикции и может найти применение в молекулярно-биологических и генно-инженерных работ.
Список литературы
- Roberts R, Belfort M, Bestor T, Bhagwat A, Bickle T, Bitinaite J, et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003;31: 1805–12. [PubMed]
- Roberts RJ. The Restriction Enzyme Database. In: The Restriction Enzyme Database [Internet]. 2019. Available: http://rebase.neb.com/rebase/link_proto
- Руководство к практическим занятиям по микробиологии. Под ред. Егорова НС. Москва: Изд-во МГУ; 1995.
- Определитель бактерий Берджи. под ред. Хоулт Д, Криг Н, Снит П, Стейли Д, Уилльямс С. Москва: Мир; 1997
- Bolivar F, Rodriguez R, Greene P, Betlach M, Heyneker H, Boyer H, et al. Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene. 1977;2: 95–113. [PubMed]
- Белавин ПА, Дедков ВС, Дегтярев СХ. Метод определения эндонуклеаз рестрикции в колониях бактерий. Прикл Биохим Микробиол. 1988;24: 121–124.
- Smith H, Nathans D. Letter: A suggested nomenclature for bacterial host modification and restriction systems and their enzymes. J Mol Biol. 1973;81: 419–23. [PubMed]
- Degtyarev S, Belichenko O, Lebedeva N, Dedkov V, Abdurashitov M. BtrI, a novel restriction endonuclease, recognises the non-palindromic sequence 5’-CACGTC(-3/-3)-3’. Nucleic Acids Res. 2000;28: E56. [PubMed]