
The restriction endonuclease EmiI, an isoschizomer of Ksp632I, recognizes the non-palindromic DNA sequence 5′-CTCTTC(1/4)-3′
M.A. Abdurashitov, D.A. Gonchar, V.A. Chernukhin, V.S. Dedkov, N.A. Mikhnenkova, A.A. Nikonova, S.Kh. Degtyarev
Authors
M.A. Abdurashitov, D.A. Gonchar, V.A. Chernukhin, V.S. Dedkov, N.A. Mikhnenkova, A.A. Nikonova, S.Kh. Degtyarev
Affiliation
SibEnzyme Ltd., Novosibirsk, Russia
* Corresponding author: M.A. Abdurashitov, SibEnzyme Ltd., 2/12 Ak. Timakova Street, Novosibirsk 630117, Russia Tel.: +7 (383) 333-4991Fax: +7 (383) 333-6853 E-mail: abd@sibenzyme.ru
Abstract
A bacterial strain, Exiguobacterium mexicanum 6, was identified as a producer of a novel restriction endonuclease designated EmiI. The enzyme recognizes the non-palindromic hexanucleotide DNA sequence 5′-CTCTTC-3′ and cleaves DNA outside the recognition site at positions 1/4, generating three-nucleotide 5′-protruding ends: 5′-CTCTTC(N)₁↓-3′ / 3′-GAGAAG(N)₄↓-5′.
Thus, EmiI is an isoschizomer of the restriction endonucleases Ksp632I [1] and Bst6I [2]. The producer strain was identified based on morphological and biochemical characteristics as well as analysis of the primary structure of a fragment of the 16S rRNA gene. A preparation of restriction endonuclease EmiI with an activity of 5000 U/ml was obtained through purification using three chromatographic steps. Optimal reaction conditions for EmiI include SE buffer Y (33 mM Tris–acetate, pH 7.9; 10 mM Mg–acetate; 66 mM potassium acetate; 1 mM DTT) at a temperature of 37 °C.
Abbreviations
BSA, bovine serum albumin; DTT, dithiothreitol; λ DNA, bacteriophage λ DNA; T7 DNA, bacteriophage T7 DNA; U, units of activity; Tris, tris(hydroxymethyl)aminomethane; PAGE, polyacrylamide gel electrophoresis; bp, base pairs; RE, restriction endonuclease; EDTA, ethylenediaminetetraacetic acid.
Keywords:
producer strain; enzyme purification; type IIS restriction endonuclease; isoschizomer.
DOI:10.26213/3034-4301.2025.7.4.002
Citation:
Abdurashitov M.A., Gonchar D.A., Chernukhin V.A., Dedkov V.S., Mikhnenkova N.A., Nikonova A.A., Degtyarev S.Kh. (2025) The restriction endonuclease EmiI, an isoschizomer of Ksp632I, recognizes the non-palindromic DNA sequence 5′-CTCTTC(1/4)-3′. DNA-Processing Enzymes, 2025, Vol. 4. DOI:10.26213/3034-4301.2025.7.4.002

This article is distributed under the terms of the Creative Commons Attribution–NonCommercial–NoDerivatives 4.0 International License (CC BY-NC-ND 4.0).
Introduction
Type IIS restriction endonucleases [3] cleave DNA at defined positions outside their recognition sites, generating unique cohesive ends, which enables their application in a range of genetic engineering and molecular biology technologies. The present study describes a novel restriction endonuclease, EmiI, which recognizes the non-palindromic hexanucleotide DNA sequence 5′-CTCTTC and cleaves DNA outside the recognition site at positions 1/4, as indicated by arrows:
5′-CTCTTC(N)₁↓-3′ / 3′-GAGAAG(N)₄↓-5′.
Experimental Conditions
Reagents used in this study were purchased from Sigma-Aldrich (USA), Fisher (Germany), Panreac (Spain), Dia-M and Helicon (Russia). The following chromatographic media were used for enzyme purification: phosphocellulose P11 and heparin–Sepharose (Sigma-Aldrich, USA), and hydroxyapatite (Bio-Rad, USA). Components of the culture media were obtained from Organotechnie (France). Restriction endonucleases, T4 polynucleotide kinase, bacteriophage λ and T7 DNA preparations, DNA molecular weight markers, and SE reaction buffers for restriction endonucleases (B100, B, G, O, W, Y, ROSE) were purchased from SibEnzyme Ltd. (Russia).
Morphological and physicochemical properties of the strain were examined using standard methods [4]. Taxonomic identification of the microorganism was performed according to Bergey’s Manual of Systematic Bacteriology [5] and based on sequencing of a PCR fragment of the 16S rRNA gene.
Assay of EmiI Restriction Activity
Bacteriophage λ DNA was used as a substrate for analysis of EmiI activity. During screening of chromatographic fractions, 1 μl aliquots from each fraction were added to 20 μl of reaction mixture containing 1 μg of λ DNA in SE buffer Y and incubated for 15 min at 37 °C.
For activity testing of the purified EmiI preparation, either 1 μl of undiluted enzyme or 1–2 μl of enzyme diluted tenfold in SE buffer B100 (10 mM Tris-HCl, pH 7.6; 50 mM KCl; 0.1 mM EDTA; 200 μg/ml BSA; 1 mM DTT; 50% glycerol) were used. The enzyme was added to 50 μl reaction mixtures containing 1 μg of λ DNA and one of six reaction buffers for restriction endonucleases. The mixtures were incubated for 1 h at 37 °C. Reaction products were analyzed by electrophoresis in a 1% agarose gel using Tris–acetate buffer (50 mM Tris–acetate, pH 8.0; 20 mM sodium acetate; 2 mM EDTA) at 180 V. After staining with ethidium bromide, gels were visualized under UV light.
One unit of restriction activity was defined as the minimum amount of enzyme required to completely digest 1 μg of λ DNA in SE buffer Y in a 50 μl reaction volume at 37 °C within 1 h.
To determine the cleavage position of EmiI within DNA, a [³²P]-labeled oligonucleotide duplex containing the EmiI recognition site and sites for control enzymes was used. Products of enzymatic cleavage were separated by electrophoresis in 20% polyacrylamide gel containing 8 M urea.
Cultivation of E. mexicanum 6 Cells
The E. mexicanum 6 strain was cultivated in a 20-liter fermenter (New Brunswick, USA) in LM broth containing 1% tryptone (Organotechnie, France), 0.5% yeast extract (same supplier), 0.5% NaCl, 0.05% MgCl₂, and 0.001% thiamine at 30 °C with agitation and aeration for 5 h until an optical density at 550 nm (OD₅₅₀) of 3.0 was reached. Cells were harvested by centrifugation using a Beckman J2-MI centrifuge (USA) with a JA-10 rotor at 8000 rpm for 20 min and stored at −20 °C.
Purification of Restriction Endonuclease EmiI
Purification conditions and buffers
All purification steps were carried out at +4 °C using the following buffers:
Buffer A: 10 mM Tris-HCl, pH 7.6; 0.1 mM EDTA; 7 mM β-mercaptoethanol.
Buffer B: 10 mM potassium phosphate, pH 7.3; 0.1 mM EDTA; 7 mM β-mercaptoethanol.
Extraction
Seventeen grams of biomass were suspended in 60 ml of buffer A containing 0.1 M NaCl, 1 mg/ml lysozyme, and 0.1 mM PMSF (phenylmethylsulfonyl fluoride). After stirring for 2 h on ice, cells were disrupted by ultrasonication at maximum power with an amplitude of 22–24 μm using a Soniprep 150 device (MSE, UK) equipped with a 2-cm probe, applying seven 60-s pulses with 2-min intervals. The lysate was clarified by centrifugation in a Beckman J2-MI centrifuge with a JA-20 rotor at 15,000 rpm for 30 min.
Phosphocellulose P11 chromatography
The clarified extract was loaded onto a 60-ml phosphocellulose P11 column equilibrated with buffer A containing 0.1 M NaCl. The column was washed with 120 ml of the same buffer. Bound proteins were eluted with a linear gradient of NaCl (0.1–0.8 M) in buffer A over a total volume of 600 ml. Sixty fractions (10 ml each) were collected. Fractions containing maximal activity (fractions 42–54) were pooled.
Hydroxyapatite chromatography
The pooled fraction was applied to an 8-ml hydroxyapatite column equilibrated with buffer B containing 50 mM NaCl and washed with 15 ml of the same buffer. The enzyme was eluted with a linear gradient of potassium phosphate (20–300 mM) in buffer B containing 50 mM NaCl over a volume of 300 ml. Fifty fractions (6 ml each) were collected. Fractions with maximal activity (fractions 24–29) were pooled and dialyzed against 2 liters of buffer A for 1.5 h.
Heparin–Sepharose chromatography
After dialysis, the pooled fraction was loaded onto a 5-ml heparin–Sepharose column equilibrated with buffer A containing 50 mM NaCl and washed with 10 ml of the same buffer. Bound proteins were eluted with a linear NaCl gradient (50–800 mM) in buffer A over a volume of 80 ml. Forty fractions (2 ml each) were collected. Fractions exhibiting maximal enzyme activity (fractions 18–23) were pooled.
Concentration and Storage of the Enzyme Preparation
BSA was added to the pooled fraction to a final concentration of 0.2 mg/ml. The preparation was then dialyzed against 200 ml of concentration buffer (10 mM Tris-HCl, pH 7.6; 0.1 mM EDTA; 7 mM β-mercaptoethanol; 0.2 M NaCl; 50% glycerol) with gentle stirring for 16 h. The enzyme preparation was stored at −20 °C.
Results and Discussion
Characterization and Taxonomic Identification of the Producer Strain
The producer strain was isolated from pond water. When cultivated on LM agar at 24 °C for 3 days, it formed circular colonies up to 4 mm in diameter. The colonies were convex to conical, shiny, opaque, and pale orange in color. Cells were rod-shaped, measuring approximately 0.6 × 3–4 μm, motile, Gram-positive, catalase-positive, and oxidase-negative. The strain grew aerobically at temperatures ranging from 8 to 40 °C but did not grow at 55 °C. The genomic DNA G+C content of the strain, determined according to [6], was 53 ± 5%.
For strain identification based on 16S rRNA gene analysis, genomic DNA was isolated from the cells, and PCR amplification of the 16S rRNA gene was performed using the primers 5′-AGAGTTTGATCMTGGCTCA-3′ and 5′-TACGGYTACCTTGTTACGACT-3′, which are modified versions of primers fD1 and rP1 [7]. A 721-bp PCR fragment corresponding to a portion of the 16S rRNA gene was sequenced (Fig. 1).
Figure 1.
Nucleotide sequence of a fragment of the 16S rRNA gene of the producer strain.
1 ATGCAGTCGA GCGCAGGACA TCGACGGAAC CCTTCGGGGG GAAGTCGACG GAATGAGCGG 61 CGGACGGGTG AGTAACACGT AAAGAACCTG CCCTCAGGTC TGGGATAACC ACGAGAAATC 121 GGGGCTAATA CCGGATGGGT CATCGAACCG CATGGTTCGA GGATGAAAGG CGCTTCGGCG 181 TCGCCTGGGG ATGGCTTTGC GGTGCATTAG CTAGTTGGTG GGGTAATGGC CCACCAAGGC 241 GACGATGCAT AGCCGACCTG AGAGGGTGAT CGGCCACACT GGGACTGAGA CACGGCCCAG 301 ACTCCTACGG GAGGCAGCAG TAGGGAATCT TCCACAATGG ACGAAAGTCT GATGGAGCAA 361 CGCCGCGTGA ACGATGAAGG CCTTCGGGTC GTAAAGTTCT GTTGTAAGGG AAGAACAAGT 421 GCCGCAGGCA ATGGCGGCAC CTTGACGGTA CCTTGCGAGA AAGCCACGGC TAACTACGTG 481 CCAGCAGCCG CGGTAATACG TAGGTGGCAA GCGTTGTCCG GAATTATTGG GCGTAAAGCG 541 CGCGCAGGCG GCCTCTTAAG TCTGATGTGA AAGCCCCCGG CTCAACCGGG GAGGGCCATT 601 GGGAAACTGG GAGGCTTGAG TATAGGAGAG AAGAGTGGAA TTCCACGTGT AGCGGTGAAA 661 TGCGTAGAGA TGTGGAGGAC ACCAGTGGCG AAGGCGACTC TTTTGGGCCT ATACTGACGC 721 TGA
Based on the determined nucleotide sequence of the 16S rRNA gene fragment, the producer strain was assigned to Exiguobacterium mexicanum of the order Bacillales, Family XII Incertae Sedis. Strain identification was performed using Nucleotide BLAST [8], the SILVAngs identification service [9], and a taxonomic classifier [10]. The microbiological characteristics of the strain were consistent with those of the assigned genus [11]. The strain was designated Exiguobacterium mexicanum 6, and the restriction endonuclease isolated from this strain was named EmiI in accordance with established nomenclature.
Preparation of EmiI and Characterization of Its Properties
Cultivation of E. mexicanum 6 cells in 20 liters of medium yielded 64 g of biomass with an EmiI activity of 50,000 U/g. Following three chromatographic purification steps, 3 ml of a restriction endonuclease EmiI preparation with a concentration of 5000 U/ml was obtained from 17 g of biomass.
Further experiments were carried out to determine the optimal reaction conditions for EmiI activity. Reaction mixtures (50 μl) contained 1 μg of bacteriophage λ DNA, one of six reaction buffers for restriction endonucleases, and 1 or 2 μl of the EmiI preparation diluted tenfold in SE buffer B100. The mixtures were incubated at 37 °C for 1 h. The results are shown in Figure 2.
Figure 2.
Activity of the EmiI preparation on bacteriophage λ DNA at 37 °C in different SE buffers (B, G, O, W, Y, and ROSE). Electrophoresis was performed in a 1% agarose gel.
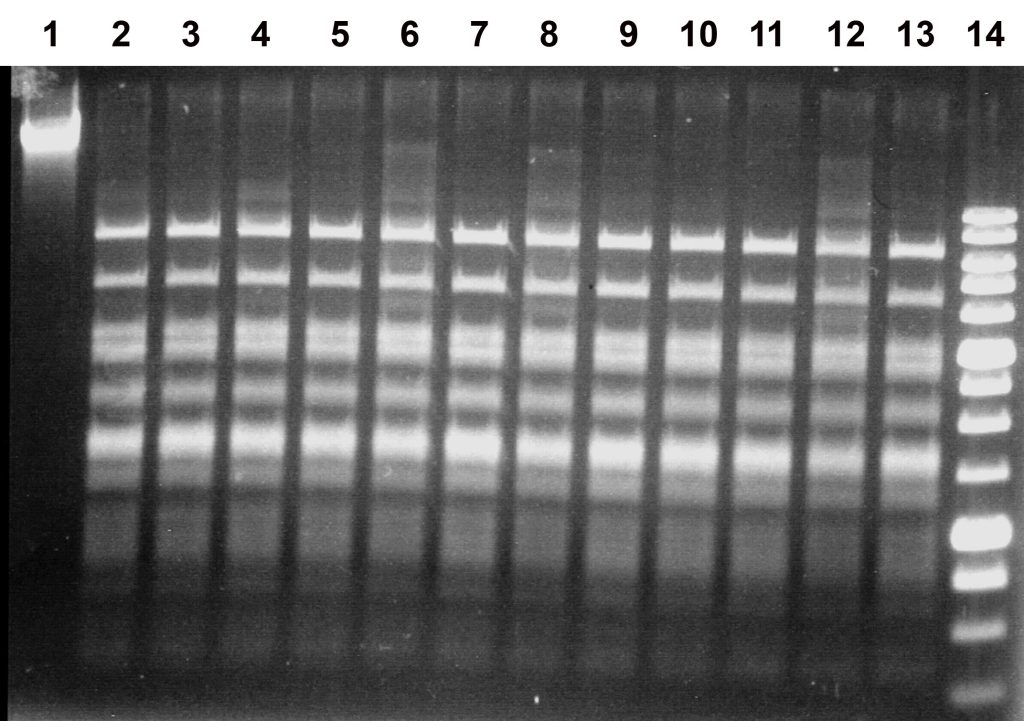
Lanes:
1, undigested λ DNA;
2, 4, 6, 8, 10, 12, λ DNA + 1 μl of EmiI diluted tenfold;
3, 5, 7, 9, 11, 13, λ DNA + 2 μl of EmiI diluted tenfold;
lanes 2–3, SE buffer B;
lanes 4–5, SE buffer G;
lanes 6–7, SE buffer O;
lanes 8–9, SE buffer W;
lanes 10–11, SE buffer Y;
lanes 12–13, SE buffer ROSE;
14, SE 1 kb DNA molecular weight marker.
As shown in Figure 2, the enzyme exhibits maximal activity in SE buffer Y (33 mM Tris–acetate, pH 7.9; 10 mM magnesium acetate; 66 mM potassium acetate; 1 mM DTT), as complete digestion of the substrate DNA was observed upon addition of both 1 and 2 μl of the tenfold-diluted enzyme preparation (lanes 10–11). In all other cases, 1 μl of the diluted EmiI preparation was insufficient for complete DNA digestion. The results of EmiI activity assays in six SE buffers at 37 °C are summarized in Table 1.
Table 1.
Activity of the restriction endonuclease preparation EmiI in six standard SE buffers for restriction endonucleases (expressed as a percentage of maximal activity).
| SE buffer | B | G | O | W | Y | ROSE |
| EmiI activity, % | 75–100 | 50–75 | 25–50 | 25–50 | 100 | 25 |
Comparison of the DNA cleavage pattern of bacteriophage λ DNA obtained using EmiI with theoretically predicted cleavage patterns based on known restriction endonuclease recognition sites led to the conclusion that EmiI is an isoschizomer of the restriction endonucleases Ksp632I [1] and Bst6I [2], which cleave this substrate at the site 5′-CTCTTC-3′ / 3′-GAGAAG-5′.
To confirm the specificity of the novel enzyme, comparative digestion of bacteriophage λ and T7 DNA with the restriction endonucleases EmiI and Bst6I was performed in 50 μl reaction mixtures containing 1 μg of DNA in SE buffer Y and 1 μl of either Bst6I or EmiI preparation. After incubation for 1 h at 65 °C (for Bst6I) or 37 °C (for EmiI), 15 μl aliquots were loaded onto a 1% agarose gel and subjected to electrophoresis. The results are shown in Figure 3.
Figure 3.
Determination of the specificity of the restriction endonuclease EmiI using bacteriophage λ and T7 DNA. Reaction products were separated in a 1% agarose gel.
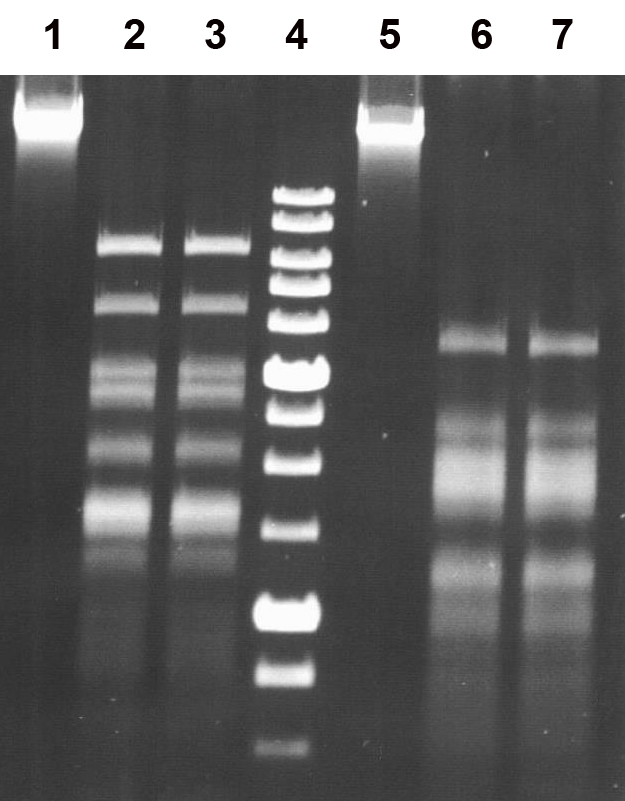
Lanes:
1, undigested bacteriophage λ DNA;
2, λ DNA + 1 μl of Bst6I preparation;
3, λ DNA + 1 μl of EmiI preparation;
5, undigested bacteriophage T7 DNA;
6, T7 DNA + 1 μl of Bst6I preparation;
7, T7 DNA + 1 μl of EmiI preparation;
4, SE 1 kb DNA molecular weight marker.
As shown in the figure, the DNA cleavage patterns produced by the restriction endonucleases Bst6I and EmiI for both DNA substrates are identical, confirming that the two enzymes share the same cleavage specificity.
To determine the optimal temperature for EmiI activity, 50 μl reaction mixtures containing 1 μg of bacteriophage λ DNA in SE buffer Y and either 1 μl of the undiluted enzyme preparation or 1 μl of the preparation diluted tenfold were incubated for 1 h at 25, 30, 37, or 55 °C. The results are presented in Figure 4.
Figure 4.
Activity of the EmiI preparation on bacteriophage λ DNA in SE buffer Y at different temperatures. Electrophoresis was performed in a 1% agarose gel.

Lanes:
1, undigested bacteriophage λ DNA;
2 and 3, λ DNA + 1 μl of undiluted or 1 μl of tenfold-diluted EmiI, 25 °C;
4 and 5, λ DNA + 1 μl of undiluted or 1 μl of tenfold-diluted EmiI, 30 °C;
6 and 7, λ DNA + 1 μl of undiluted or 1 μl of tenfold-diluted EmiI, 37 °C;
8 and 9, λ DNA + 1 μl of undiluted or 1 μl of tenfold-diluted EmiI, 55 °C;
10, SE 1 kb DNA molecular weight marker.
As shown in Figure 4, complete digestion of the substrate DNA by 1 μl of the undiluted EmiI preparation was observed at all tested temperatures (lanes 2, 4, 6, and 8). In contrast, when the enzyme was diluted tenfold, exhaustive digestion of the substrate was observed only at 37 °C (lane 7). Therefore, this temperature was determined to be optimal for EmiI restriction activity. Complete inactivation of EmiI was observed after heating the reaction mixture to 80 °C for 20 min (data not shown).
Based on these results, the activity of the EmiI preparation was determined. Figure 5 shows an electropherogram illustrating analysis of the target enzyme activity at the optimal temperature (37 °C) in the optimal SE buffer Y.
Figure 5.
Determination of EmiI activity under optimal conditions. Reaction mixtures (50 μl) contained 1 μg of bacteriophage λ DNA in SE buffer Y and either 1 μl of undiluted EmiI (control) or 0.5, 1, or 2 μl of EmiI diluted tenfold in buffer B100. The mixtures were incubated for 1 h at 37 °C and analyzed by electrophoresis in a 1% agarose gel.

Lanes:
1, undigested bacteriophage λ DNA;
2, λ DNA + 0.5 μl of EmiI diluted tenfold;
3, λ DNA + 1 μl of EmiI diluted tenfold;
4, λ DNA + 2 μl of EmiI diluted tenfold;
5, λ DNA + 1 μl of undiluted EmiI;
6, SE 1 kb DNA molecular weight marker.
As shown in Figure 5, exhaustive digestion of bacteriophage λ DNA by the restriction endonuclease EmiI was observed upon addition of either 2 μl of the enzyme preparation diluted tenfold (lane 4) or 1 μl of the undiluted enzyme preparation (lane 5). Thus, the activity of the EmiI preparation was determined to be 5 U/μl (5000 U/ml). One unit of EmiI restriction activity was defined as the minimum amount of enzyme required for complete digestion of 1 μg of bacteriophage λ DNA in a 50 μl reaction mixture within 1 h in SE buffer Y at 37 °C.
To confirm the recognition site of EmiI and to determine the position of DNA cleavage by the new enzyme, two 5′-end radiolabeled oligonucleotide duplexes, Emi1*/Emi2 and Emi2*/Emi1 (the labeled strand is indicated by an asterisk), were used. These duplexes were formed from two oligonucleotides containing the EmiI recognition site (underlined):
Emi1: 5′-CCTTTGACGCTCTTCGGCGCCATGATGCAACC-3′
Emi2: 5′-GGTTGCATCATGGCGCCGAAGAGCGTCAAAGG-3′
The duplexes were digested with the novel enzyme EmiI and, separately, with control restriction endonucleases Bst6I (5′-CTCTTC(1/4)-3′), HgaI (5′-GACGC(5/10)-3′), HspAI (5′-G↓CGC-3′), FaeI (5′-CATG↓-3′), and SfaNI (5′-GCATC(5/9)-3′). Reactions were carried out using 1 pmol of the Emi1*/Emi2 and Emi2*/Emi1 duplexes in a 10 μl reaction volume for 1 h at 37 °C (or at 55 °C for Bst6I) in SE buffer Y. The cleavage products were separated by electrophoresis in a 20% polyacrylamide gel containing 8 M urea using Tris–borate buffer. An autoradiograph of the gel after electrophoresis is shown in Figure 6A.
Figure 6A.
Determination of the EmiI cleavage position. Electrophoresis was performed in a 20% polyacrylamide gel containing 8 M urea.
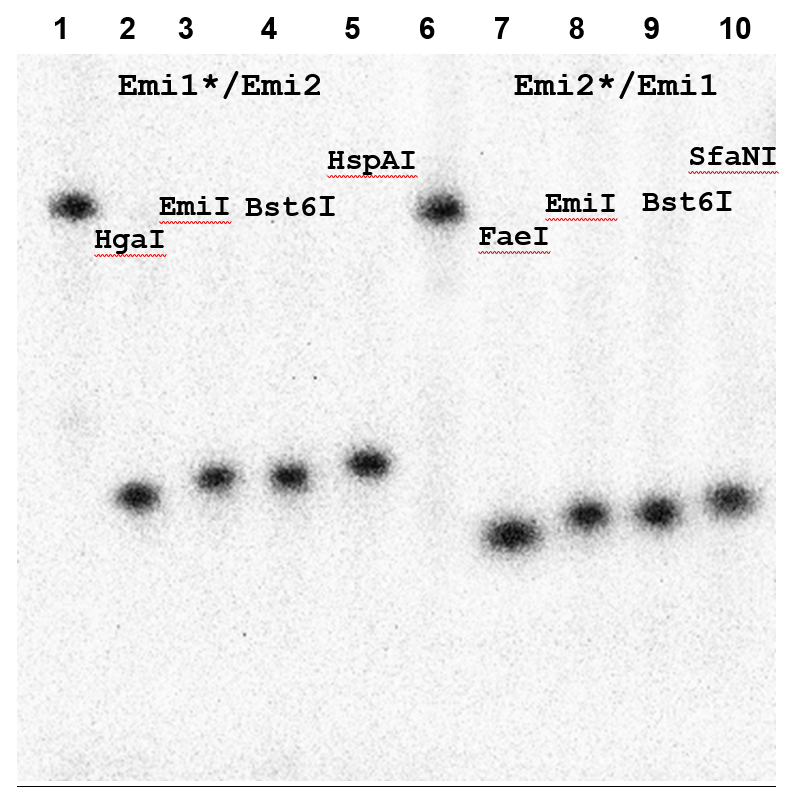
Lanes:
1, untreated Emi1*/Emi2 duplex;
2, Emi1*/Emi2 duplex + 1 μl of HgaI;
3, Emi1*/Emi2 duplex + 1 μl of EmiI;
4, Emi1*/Emi2 duplex + 1 μl of Bst6I;
5, Emi1*/Emi2 duplex + 1 μl of HspAI;
6, untreated Emi2*/Emi1 duplex;
7, Emi2*/Emi1 duplex + 1 μl of FaeI;
8, Emi2*/Emi1 duplex + 1 μl of EmiI;
9, Emi2*/Emi1 duplex + 1 μl of Bst6I;
10, Emi2*/Emi1 duplex + 1 μl of SfaNI.
Figure 6B shows the structure of the Emi1/Emi2 duplex with the recognition sites and cleavage positions of the restriction endonucleases used for analysis indicated. For clarity, the recognition sites and arrows denoting cleavage positions for each restriction endonuclease are highlighted in different colors. For each strand of the duplex, the enzymes were selected such that their cleavage positions either coincided with the predicted EmiI cleavage position (as in the case of Bst6I) or differed by one nucleotide from the 5′ end (HgaI for the upper strand and FaeI for the lower strand) or by one nucleotide from the 3′ end (HspAI for the upper strand and SfaNI for the lower strand).
Figure 6B.
Schematic representation of the Emi1/Emi2 oligonucleotide duplex indicating the recognition sites and cleavage positions of the control restriction endonucleases.

The data presented in Figure 6A indicate that the EmiI cleavage position coincides with that of Bst6I (5′-CTCTTCN↓-3′ / 3′-GAGAAGNNNN↓-5′) and differs by one nucleotide in either direction from the cleavage positions of the control enzymes.
Thus, the restriction endonuclease EmiI from Exiguobacterium mexicanum 6 recognizes the non-palindromic hexanucleotide DNA sequence 5′-CTCTTC-3′ and cleaves DNA outside the recognition site at positions 5′-CTCTTC(N)₁↓-3′ / 3′-GAGAAG(N)₄↓-5′, generating three-nucleotide 5′-protruding ends, similarly to the previously described restriction endonucleases Ksp632I and Bst6I. The novel enzyme may be applied in research in the fields of genetic engineering, molecular biology, biotechnology, and DNA diagnostics.
References
-
-
Bolton, B.J., Schmitz, G.G., Jarsch, M., Comer, M.J., Kessler, C.
Ksp632I, a novel class IIS restriction endonuclease from Kluyvera sp. strain 632 with the asymmetric hexanucleotide recognition sequence 5′-CTCTTC(N)₁-3′ / 3′-GAGAAG(N)₄-5′.
Gene 66(1) (1988) 31–43.
DOI: 10.1016/0378-1119(88)90222-3. -
Restriction endonuclease Bst6I.
SibEnzyme product page.
Available at: http://sibenzyme.com/product/bst6-i/ -
Pingoud, A., Fuxreiter, M., Pingoud, V., Wende, W.
Type II restriction endonucleases: structure and mechanism.
Cellular and Molecular Life Sciences 62 (2005) 685–707.
DOI: 10.1007/s00018-004-4513-1. -
Egorov, N.S. (Ed.).
Manual for Practical Classes in Microbiology.
Moscow, 1995. (in Russian) -
Holt, J.G. et al. (Eds.).
Bergey’s Manual of Determinative Bacteriology. 9th ed., 2 vols.
Russian translation edited by G.A. Zavarzin.
Moscow, 1997. -
Dedkov, V.S.
Determination of G+C content in bacterial DNA using restriction endonucleases.
Biotekhnologiya (Biotechnology) 4 (2004) 77–82. (in Russian) -
Weisburg, W.G., Barns, S.M., Pelletier, D.A., Lane, D.J.
16S ribosomal DNA amplification for phylogenetic study.
Journal of Bacteriology 173 (1991) 697–703.
DOI: 10.1128/jb.173.2.697-703.1991. -
Nucleotide BLAST.
National Center for Biotechnology Information.
Available at: https://blast.ncbi.nlm.nih.gov -
SILVAngs.
SILVA rRNA gene alignment and classification service.
Available at: https://www.arb-silva.de/aligner -
NCBI Taxonomy Browser.
Available at: https://www.ncbi.nlm.nih.gov/Taxonomy/ -
Raichand, R., Pareek, S., Singh, N.K., Mayilraj, S.
Exiguobacterium aquaticum sp. nov., a member of the genus Exiguobacterium.
International Journal of Systematic and Evolutionary Microbiology 62 (2012) 2150–2155.
DOI: 10.1099/ijs.0.035790-0.
-